

Research
We study the molecular genetics of blood clotting and the structure, function, and cellular production of blood clotting and other proteins.

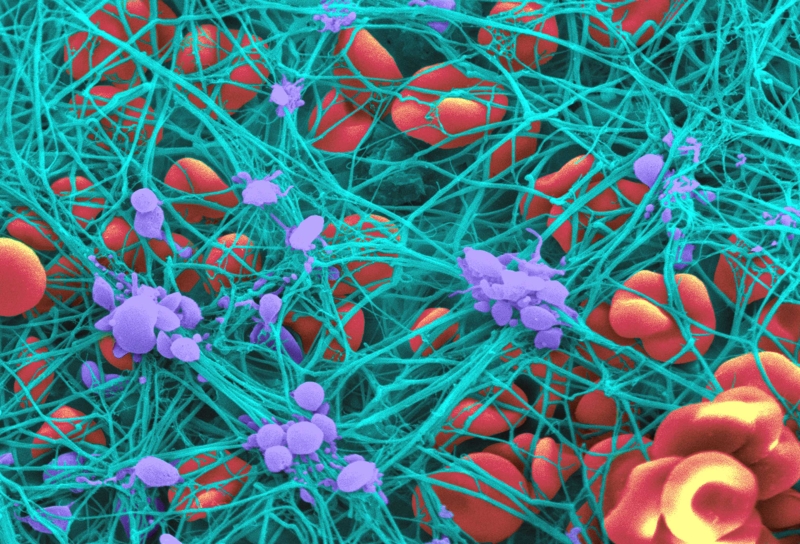
Precise control of the blood-clotting system is essential for maintenance of the circulation in all higher animals. Deficient function of this system can lead to fatal bleeding following even a minor injury, whereas overactivity of this system can produce unwanted blood clots, resulting in blockages to critical blood vessels, as occurs in such diseases as heart attack and stroke.
The Ginsburg lab has a longstanding interest in the genetics of inherited bleeding and thrombotic disorders and the structure and function of blood coagulation proteins, including von Willebrand factor (von Willebrand disease), ADAMTS13 (TTP), and PAI-1 (PAI-1 deficiency). Our discovery of the molecular basis for combined deficiency of factors V and VIII took us from the “bedside” to the “bench” to further explore the regulation of protein transport from the endoplasmic reticulum to the Golgi, leading to new insights into the pathogenesis of heart and blood diseases, including the control of cholesterol levels.
The lab also studies the heterogeneity of endothelial cell gene expression and the role of these cells in regulating hemostasis and the vascular response to inflammation.
Current Projects include: