


Scientists develop greener, more efficient method for producing next-generation antibiotics
An international team of researchers has developed a method for altering one class of antibiotics, using microscopic organisms that produce these compounds naturally. The findings, published July 25 in Nature Chemistry, could lead to more efficient production of antibiotics that are effective against drug-resistant bacteria.
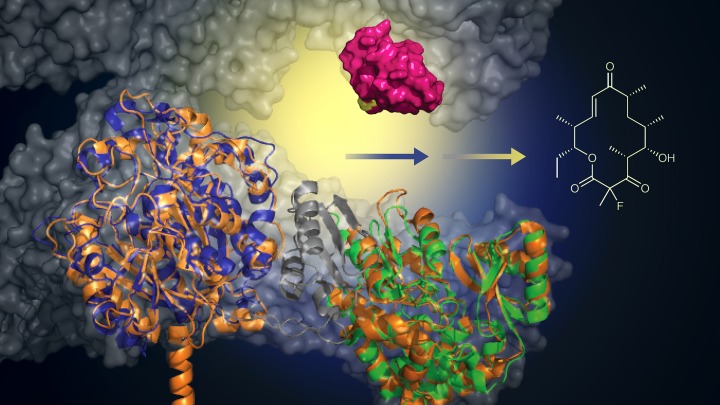
The team started with a microorganism that is genetically programmed to produce the antibiotic erythromycin. Scientists from the Institute of Organic Chemistry and Chemical Biology at Germany’s Goethe University wondered if the system could be genetically altered to assemble the antibiotic with one additional fluorine atom, which can often improve pharmaceutical properties.
“We had been analyzing fatty acid synthesis for several years when we identified a part of a mouse protein that we believed could be used for directed biosynthesis of these modified antibiotics, if added to a biological system that can already make the native compound,” says Martin Grininger, Ph.D., professor for biomolecular chemistry at Goethe University.
Working with the lab of David Sherman, Ph.D., at the University of Michigan, which specializes in this biological assembly system, the team used protein engineering to replace one part of the system’s native machinery with the functionally similar mouse gene.
“It’s like taking one engine part out of a Mercedes and putting it into a Porsche to make a better hybrid engine. You get a Porsche engine that can do new things and works even better,” says Sherman, a faculty member at the U-M Life Sciences Institute and professor of medicinal chemistry in the College of Pharmacy. “We can now take advantage of this protein engineering to make new compounds that have this very desirable fluorine atom, which chemists have been struggling to add to macrolide antibiotics for a long time.”
Why fluorine?
The reason this added fluorine atom is so desirable is that it changes not only the structure of the final product, but the product’s ability to kill bacteria and work safely in patients.
Erythromycin works by binding to and blocking the activity of the bacterial ribosome, which is essential for bacteria to survive. Some bacteria have evolved ways to prevent this binding, making them resistant to treatment with antibiotics. Altering the antibiotic’s structure with a fluorine atom overcomes that evolutionary advantage, restoring the compound’s ability to fight bacteria.
While chemists have developed methods for adding the fluorine synthetically, the process is arduous and requires the use of toxic chemical reagents. The new biosynthetic method developed by the researchers from Goethe University and U-M overcomes those challenges.
“It’s a very exciting development, because we can bypass all the time-consuming synthetic steps and dangerous chemicals,” Sherman explains. “We have shown that we can basically reprogram an organism to make the fluorinated product directly.”
The researchers emphasize that the fluorinated compounds are still a few years away from being available in the clinic. But the findings offer a more efficient path forward for developing new antibiotics, and even antivirals and anti-cancer medications.
“Our approach has been proven successful on a small set of antibiotics, but it could ultimately be used to develop a wide range of pharmaceuticals with minimal use of toxic chemicals and by-products,” Grininger says.
Top image: With the addition of a murine-derived biocatalyst (green), this engineered protein can add a fluoride atom to create macrolide analogs (structure, right). This approach offers a greener, more efficient method for creating new antibiotics. Image courtesy of Martin Grininger and Rajani Arora.
Go to Article
“Chemoenzymatic synthesis of fluorinated polyketides,” Nature Chemistry. DOI: 10.1038/s41557-022-00996-z